In 2003, the International Human Genome Sequencing Consortium completed the first whole genome sequencing of human DNA after years of worldwide collaboration and billions of dollars of investment. Many human genomes have since been sequenced, the analysis of which has lead to many important discoveries. The factors that contribute to disease and if they affect different populations in different ways can be discovered by determining the correlations between people’s lifestyle, family history, environment and genomic data. There is now widespread motivation to sequence the genomes of hundreds of thousands, even millions, of individuals to learn even more. The global market for genomics is expected to reach USD 23.88b by 2022 (up from $14.71b in 2017), growing at an estimated CAGR of 10.2% from 2017 to 2020 due to increased investment in diagnostic services.
A genome is the complete set of DNA found in the cell of an organism. To perform a genetic analysis on a human population the individual genomes must each be digitized. The first step is to extract all the DNA from an individual’s cell so it can be read by a DNA reader. Due to technological limitations the DNA strands must be sliced into short strands for the reader to operate. The reader can then perform reads and the resulting sequences of G’s, T’s, C’s and A’s are written to file in a format known as FASTQ. During this process the original DNA ordering is lost. Consequently, the second step is to align the reads in the FASTQ file against a reference genome so the original order of the individual’s DNA can be recovered. The alignment program writes reads and their position in the reference genome to a file in a format known as SAM.
To perform studies on a massive scale, biologists need to be able to align thousands of genomes per hour. Right now sequencing is conducted using state-of-the-art aligners such as Bowtie2 and BWA. These are excellent tools for aligning thousands of genomes per week and have good support for multiple-cores, but few are built with the scenario of aligning thousands of genomes per hour in mind. In order do so, one needs to make use of multiple machines. Tools such as pMap and mpiBWA support multiple machines (both using MPI), but have their own issues. To build a highly scalable alignment pipeline the following issues have to addressed:
- Filesystem: Every machine in the cluster needs access to the indexed referenced genome, which may be several gigabytes in size. Current shared filesystems perform extremely poorly when concurrently accessed by many machines and may become a significant bottleneck and point of failure. Such issues are commonly encountered by anyone attempting to run Python on large clusters.
- Throughput: Critical to maximizing throughput is the concept that the only time a machine is idle is when it is limited by an unavoidable bottleneck. In order to maximize throughput, it should be ensured that reads to be aligned are sent to worker machines as quickly as possible.
- Load balancing: As the number of machines increase that we wish to distribute across, load balancing becomes increasingly important. Reads take different amounts of time to align, and some machines may perform slower than others (e.g. due to a less powerful CPU or lower network bandwidth). The scenario where the majority of machines in cluster are idle because the final few have a disproportionate amount of work should be avoided. Tools such as pMap statically decide the amount of work to allocate to each node, which can lead to long tails in execution time.
This additional engineering would shift biologists’ focus away from actual research to building a solution that works at scale. Furthermore, much of the genome analysis completed to date cannot be combined rigorously because the different studies are separated in time to such an extent that they have been performed with slightly different versions of the aligner and reference genome. Additionally, due to the time already invested in aligning against older versions, the newest and best version of the reference genome is seldom used.
At Hadean we wondered whether we could build a sequence aligner that matches the performance of state-of-the-art tools, but with the advantage of being completely scalable and not requiring any devops skills; one that biologists could use with ease.
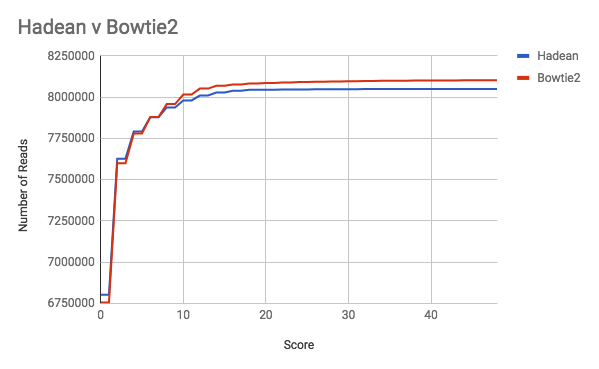
In the graph below we see Bowtie2 and our Hadean prototype perform very similarly. The graph shows the number of reads aligned with a particular score, where the score is a measure of how many changes have to be made to a read in order for it to be aligned somewhere in the reference genome. A score of zero means no changes were necessary, while a high score means that many changes were necessary.
Imagine we have a batch of FASTQ files residing on remote storage. We want to align the reads in these files and write the SAM files to the same location. The shell command to run Bowtie2 is:
bowtie2-align -p $n -x <reference_genome> { <fastq1.fq>, …, <fastqk.fq> } -S <sam_file>
Using $n thread we can align one or multiple FASTQ files. To scale this command biologists would need to develop in-depth knowledge of shell scripting, remote storage APIs, cloud APIs and networking to address the issues raised earlier. Using Bowtie2 to scale would therefore come at a price — either biologists sacrifice man-years of research time and grant money or they outsource the solution development to professionals. A standard team of eight developers costs $800K per year. If we estimate scaling Bowtie2 with a team this size would take roughly 6 months, and add in a margin for error and time spent by the biologists giving input, we arrive at an estimated cost of $500K. Beyond this, once the solution is built there would be running costs (e.g. a typical analytics solution processing 3TB costs roughly $180K in infrastructure per month).
The shell command to run the alignment on Hadean is:
cargo run — bin coordinator <reference_genome> <batch_directory> <remote_bucket>
No additional devops is required at all and it can run against an arbitrary number of genomes by just supplying the file locations. The Hadean aligner addresses the issues raised earlier in the following ways:
- Filesystem: We use an efficient broadcast mechanism which enables multiple machines to receive a copy of the genome. Since what data is sent and when is under control of the broadcasting node, there is no possibility for overload (which occurs when many machines attempt to make connections to a shared filesystem).
- Throughput: We have built abstractions in Hadean that enable messages to be dispatched to one of a set of machines based on metrics such as network buffer occupancy. In MPI we’d need to manually keep track of how many asynchronous sends were in progress and their sizes to be able to estimate how much data was buffered.
- Load balancing: We choose which machine to dispatch a read to based on network buffer occupancy and therefore achieve dynamic load balancing which transparently adapts to any changes in machine performance or network behaviour. The number of machines spawned by Hadean is proportional to the amount of FASTQ data to be aligned, which means the batch is completed in almost constant time.
- File I/O: The align program is smart enough to write the SAM files to remote storage with a file directory structure that mirrors the batch file directory.
Using Hadean we’ve demonstrated how the number of genomes analysed in a single batch is only limited by the cloud computing budget of the user. We’ve removed the technical barriers to updating studies so they are based on the newest reference genome and give biologists the ability to update studies involving hundreds of thousands of genomes in a matter of days!